The FDA generally requires manufacturers to demonstrate that a new medical device is safe and effective, i.e., substantially equivalent (SE), to an already legally marketed device via a premarket submission (a 510(k)). This is done by comparing the subject device to one (or more) legally marketed device(s), the “predicate(s)”. Sometimes there are not equivalent devices; in those cases, a De Novo submission may be required. Class III devices (the highest risk) require an application for Premarket Approval (PMA). Still, most devices that require a submission can use a 510(k).
The remainder of this article will discuss submissions in which there is a predicate device and a 510(k) is appropriate. For more information on De Novo products see the following link: https://www.fda.gov/medical-devices/premarket-submissions/de-novo-classification-request. For more information on Premarket Approvals (PMAs) see the following link: https://www.fda.gov/medical-devices/premarket-submissions/premarket-approval-pma
To establish substantial equivalence (SE), the manufacturer must compare the critical characteristics of the subject device to the critical characteristics of the predicate. For SE to be demonstrated, the following conditions must be met:
- The subject device must have the same intended use as the predicate; and
- The subject device must have the same technological characteristics as the predicate.
OR
- The subject device must have the same intended use as the predicate; and
- Any technological characteristics that are different do not raise questions of safety and effectiveness; and
- Information is provided to demonstrate the subject device is as safe and effective as the predicate.
Whether or not you are required to submit a 510(k) will depend on how the FDA’s three-level risk classification applies to your device.
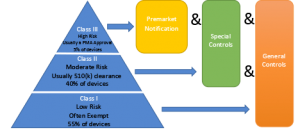
So, who is required to submit a 510(K)?
- Manufacturers (domestic and foreign) introducing a new device:
- If you make the finished device
- If you contract someone else to make the device
- Note: Accessories sold to the end user are considered medical devices
- If you repack a device and repackaging significantly changes or affects the device
- Changes in sterilization
- If you relabel a label a device and the changes are significant
- Changes to the intended use
- Changes to warnings, contraindications, etc.
Who is not required to submit a 510(k)?
- If your unfinished device is sold to another firm for further processing or assembling.
- You cannot sell to an end user.
- If you are not marketing or commercially distributing your device.
- If you distribute another firm’s manufactured device (that already has clearance).
- Must label as “Distributed by ABC Firm” or “Manufactured for ABC Firm.”
- If you repackage or relabel and the existing label or condition of the device is not significantly changed.
- Labeling should be consistent with the labeling submitted in the 510(k) with the same indications for use and warnings and contraindications.
- If your device was legally in commercial distribution before May 28, 1976, and has not been significantly changed or modified in design, components, method of manufacture, or intended use.
- If your device is made outside of the US and you are an importer of the device. The 510(k) needs to have been submitted by the manufacturer.
- If your device is exempt from 510(k) regulation
- See 21 CFR 862-892
- Certain Class I or II devices
Even if you are not required to submit a 510(k), i.e., the device is exempt, there are requirements such as establishment registration and device listing that remain applicable. For Class II, Class III, and select Class I devices, manufacturers are required to use design controls that are established within their Quality Management System (QMS) during the development of their devices. A substantive review of a submission is as much a review of a QMS as a determination of SE.
How do you submit to the FDA? What can be expected in the process?
- Log-in and acknowledgement procedure
- The FDA no longer accepts paper copies of submissions; all submissions must be electronic, whether transmitted electronically (as an “eSubmission”) or mailed to FDA on CD, DVD, or flash drive (as an “eCopy”). Additionally, the FDA only accepts submissions in the approved formatting. For additional information see the following links:
- Webpage link to the FDA guidance eCopy Program for Medical Device Submissions: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/ecopy-program-medical-device-submissions
- FDA webpage for the eCopy Program for Medical Device Submissions: https://www.fda.gov/medical-devices/how-study-and-market-your-device/ecopy-program-medical-device-submissions
- The FDA verifies that the fee payment was received.
- Pro Tip: While you can mail in a check to the FDA, the easiest way is to pay via the MDUFA webpage. Recommend doing this about 1-2 weeks prior to submission date. You will receive a K# at this time if you do this process.
- The FDA confirms a valid copy of the 510(k) was provided. For this step, a valid copy includes:
- The 510(k) is in the correct format (per FDA definition).
- Pro Tip: Run your submission through the FDA’s eSubmitter software to confirm your formatting is correct.
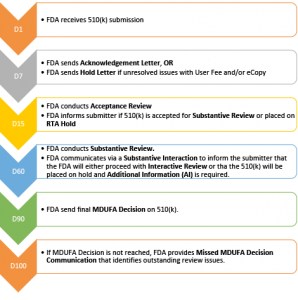
- Submitter receives an Acknowledgement Letter if submission meets all basic requirements; if not, submitter receives a Hold Letter.
- Date on this letter becomes D1 of the FDA’s clock
- FDA provides submitter with a K# if you do not already have one.
- The 510(k) is in the correct format (per FDA definition).
- The FDA no longer accepts paper copies of submissions; all submissions must be electronic, whether transmitted electronically (as an “eSubmission”) or mailed to FDA on CD, DVD, or flash drive (as an “eCopy”). Additionally, the FDA only accepts submissions in the approved formatting. For additional information see the following links:
- This K# should be on all future communications.
- This typically occurs within 7 days of receipt of the 510(k).
- The FDA performs an Acceptance Review, commonly known as an RTA Review (Refuse to Accept Review). For additional information see the following links:
- Webpage link to the FDA guidance Refuse to Accept Policy for 510(k)s: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/refuse-accept-policy-510ks
- Webpage link to the FDA ‘s webpage for Acceptance Checklists for 510(k)s: https://www.fda.gov/medical-devices/premarket-notification-510k/acceptance-checklists-510ks
- The FDA determines if the 510(k) meets the minimum threshold of acceptability to proceed to the substantive review.
- The submitter should receive an Acknowledgement Letter within 15 days of the submission.
- An Acceptance Letter informs the submitter of the lead reviewer assigned to the 510(k) and their contact information.
- The Acceptance Letter indicates the status of the 510(k). The three possibilities are:
- The 510(k) was accepted for substantive review.
- The 510(k) was not accepted for review (“refused to accept” or “RTA”)
- The submission is then placed on an RTA Hold.
- The submitter has 180 days to fully address the deficiencies cited in hold letter.
- After 180 days, if the submitter has not addressed the deficiencies the submission is considered withdrawn and deleted from FDA system.
- The 510(k) is under substantive review due to the reviewer not completing the acceptance review within 15 days.
- Substantive Review (including Substantive Interaction and Interactive Review)
- The review should occur within 60 days of D1 (Acknowledgement letter date).
- Interactive Review: typically, an email, but could be a phone call, stating that the FDA will proceed to resolve any outstanding deficiencies in which the “FDA clock” does not stop.
- The FDA reviewer will consider this if they believe the deficiencies can be resolved within the timeline established by Congress.
- Additional Information (AI):
- This places the submission on hold.
- The submitter has 180 days from the date of the AI Request to submit a complete response; after 180 days the submission is considered withdrawn.
- Responses should include the following:
- The submitter’s name
- The 510(k) number
- Identify the submission as an AI to the 510(k)
- A list of the date(s) of FDA’s request(s) for AI
- Provide the requested information in an organized manner.
- For additional information on the FDA’s review clock and goals or communicating with the FDA see the following links:
- Webpage link to the FDA ‘s webpage for FDA and Industry Actions on Premarket Notification (510(k)) Submissions: Effect on FDA Review Clock and Goals: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/fda-and-industry-actions-premarket-notification-510k-submissions-effect-fda-review-clock-and-goals
- Webpage link to the FDA ‘s webpage for Types of Communication During the Review of Medical Device Submissions: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/types-communication-during-review-medical-device-submissions
- 510(k) Decision Letter
- The FDA’s MDUFA goal to reach a decision on a 510(k) is 90 FDA Days. FDA Days exclude days the submission was on hold waiting for a response to AIs.
- The submitter will receive a decision letter via email.
- Letters that receive an SE (Substantial Equivalent) decision are considered “cleared” by the FDA. The IFU will typically be attached as well and are also considered cleared by the FDA.
- The 510(k) is added to the 510(k) database
- If no decision is reached within 100 FDA Days, the FDA will issue a Missed MDUFA Communication letter.
-
- The FDA will request that a meeting or teleconference be set up to discuss major outstanding issues or any other reasons that are preventing the FDA from reaching a final decision, with an estimated completion date.
How long will clearance take?
This is the $64,000 question! To better understand the answer, we need to understand “FDA Days” a bit more. Below is the classic chart used in RA departments around the globe to tell their teams when FDA clearance will be obtained.
However, if the request for additional information is for a sterilization validation or a complex animal study and you have not performed the testing (or even started the testing), the turnaround time can be months. If there are multiple complex questions, this can even lead to a withdrawal of the submission. Any of these scenarios may lead to significant delays and a timeline that is significantly harder to predict.
So, when pressed for an approval date, the best we can do is to fall back on the averages. The FDA sees thousands of 510(k) submissions per year, and they average 180 days for a review, with only about 19% gaining clearance within 3 months. Having an experienced team assessing the testing and submission strategy, navigating the FDA forms and communications, as well as writing the submission and answering the FDAs questions can help reduce the time and prepare cross-functional teams for the road ahead.