Discover the De Novo Review process with Fang Consulting – your trusted medical device regulatory affairs partner.
DE NOVO CLASSIFICATION

Discover the De Novo Review process with Fang Consulting – your trusted medical device regulatory affairs partner. Learn what makes a De Novo Review different from a 510(k) and PMA and how we can assist you in obtaining market authorization for your innovative medical devices.
What is a De Novo Review?
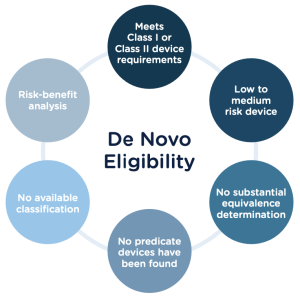
A De Novo Review is a regulatory pathway for medical devices that do not have a legally marketed predicate device to gain market authorization through the US Food and Drug Administration (FDA). In other words, if a medical device is novel and does not have a substantially equivalent device already on the market, it can be reviewed under the De Novo pathway.
The De Novo pathway was created in 1997 by the FDA Modernization Act, which amended the Federal Food, Drug, and Cosmetic Act. The De Novo pathway is intended to provide an efficient and predictable path for novel medical devices to reach the market while ensuring they are safe and effective.
What is a DEN number?
When a medical device is cleared through the De Novo pathway, it is assigned a unique identifier called a De Novo number or DEN. This number helps the FDA track the device and allows other companies to reference the device as a predicate for future 510(k) submissions.
How is a De Novo Review different from a 510(k) and PMA?
A 510(k) submission is a premarket notification for specific medical devices that demonstrate substantial equivalence to a legally marketed predicate device not subject to premarket approval. The 510(k) pathway is designed for medical devices similar to devices already on the market and has a low to moderate risk to the patient.
In contrast, a De Novo Review is intended for devices without a predicate on the market or classified as Class III devices. Class III devices support or sustain human life, are of substantial importance in preventing impairment of human health or present a potential, unreasonable risk of illness or injury.
The PMA pathway is the most stringent regulatory pathway for medical devices and is required for devices that are considered high-risk or novel. The PMA pathway requires a comprehensive submission of clinical data and is subject to extensive FDA review before approval.
The De Novo pathway provides an alternative to the PMA pathway for devices that do not have a predicate device on the market. The De Novo pathway allows the FDA to review the safety and effectiveness of the device and assign it to Class I or Class II. Once a device is classified as a class II and has a predicate, the device can be cleared through the 510(k) pathway.
What are the challenges of filing a De Novo?
While the De Novo pathway provides an alternative to the PMA pathway, there are some drawbacks to filing a De Novo. One of the main drawbacks is that navigating the process can be confusing and time-consuming, especially for companies new to the medical device industry.
Another potential challenge is that once a device clears through the De Novo pathway, a competitor can use it as a predicate device for a 510(k) submission, creating competition for the De Novo device manufacturer and making it more challenging to maintain market share.
Choose Fang Consulting to Navigate the De Novo Review Process
Fang Consulting has extensive experience helping medical device companies navigate the De Novo Review process. Our team of experts can assist with preparing and submitting the necessary documentation, including clinical data, bench testing, and risk assessments.
Our regulatory affairs professionals can also help clients develop a comprehensive regulatory strategy, which includes identifying the most appropriate pathway for their device. We understand that navigating the regulatory landscape can be challenging, and we are committed to providing our clients with the guidance and support they need to succeed.
In addition to our regulatory expertise, Fang Consulting also provides a range of other services to support medical device companies, including product development, quality management, and post-market surveillance. We work closely with our clients to ensure that their devices meet regulatory requirements and are safe and effective for patients.
If you are considering filing a De Novo Review for your medical device or need assistance with any aspect of the regulatory process, please get in touch with Fang Consulting to learn more about our services and how we can help you bring your device to market.
DE NOVO CLASSIFICATION
Discover the De Novo Review process with Fang Consulting – your trusted medical device regulatory affairs partner.

How can we help?
Discover the De Novo Review process with Fang Consulting – your trusted medical device regulatory affairs partner. Learn what makes a De Novo Review different from a 510(k) and PMA and how we can assist you in obtaining market authorization for your innovative medical devices.
What is a De Novo Review?
A De Novo Review is a regulatory pathway for medical devices that do not have a legally marketed predicate device to gain market authorization through the US Food and Drug Administration (FDA). In other words, if a medical device is novel and does not have a substantially equivalent device already on the market, it can be reviewed under the De Novo pathway.
The De Novo pathway was created in 1997 by the FDA Modernization Act, which amended the Federal Food, Drug, and Cosmetic Act. The De Novo pathway is intended to provide an efficient and predictable path for novel medical devices to reach the market while ensuring they are safe and effective.
What is a DEN number?
When a medical device is cleared through the De Novo pathway, it is assigned a unique identifier called a De Novo number or DEN. This number helps the FDA track the device and allows other companies to reference the device as a predicate for future 510(k) submissions.
How is a De Novo Review different from a 510(k) and PMA?
A 510(k) submission is a premarket notification for specific medical devices that demonstrate substantial equivalence to a legally marketed predicate device not subject to premarket approval. The 510(k) pathway is designed for medical devices similar to devices already on the market and has a low to moderate risk to the patient.
In contrast, a De Novo Review is intended for devices without a predicate on the market or classified as Class III devices. Class III devices support or sustain human life, are of substantial importance in preventing impairment of human health or present a potential, unreasonable risk of illness or injury.
The PMA pathway is the most stringent regulatory pathway for medical devices and is required for devices that are considered high-risk or novel. The PMA pathway requires a comprehensive submission of clinical data and is subject to extensive FDA review before approval.
The De Novo pathway provides an alternative to the PMA pathway for devices that do not have a predicate device on the market. The De Novo pathway allows the FDA to review the safety and effectiveness of the device and assign it to Class I or Class II. Once a device is classified as a class II and has a predicate, the device can be cleared through the 510(k) pathway.
What are the challenges of filing a De Novo?
While the De Novo pathway provides an alternative to the PMA pathway, there are some drawbacks to filing a De Novo. One of the main drawbacks is that navigating the process can be confusing and time-consuming, especially for companies new to the medical device industry.
Another potential challenge is that once a device clears through the De Novo pathway, a competitor can use it as a predicate device for a 510(k) submission, creating competition for the De Novo device manufacturer and making it more challenging to maintain market share.
Choose Fang Consulting to Navigate the De Novo Review Process
Fang Consulting has extensive experience helping medical device companies navigate the De Novo Review process. Our team of experts can assist with preparing and submitting the necessary documentation, including clinical data, bench testing, and risk assessments.
Our regulatory affairs professionals can also help clients develop a comprehensive regulatory strategy, which includes identifying the most appropriate pathway for their device. We understand that navigating the regulatory landscape can be challenging, and we are committed to providing our clients with the guidance and support they need to succeed.
In addition to our regulatory expertise, Fang Consulting also provides a range of other services to support medical device companies, including product development, quality management, and post-market surveillance. We work closely with our clients to ensure that their devices meet regulatory requirements and are safe and effective for patients.
If you are considering filing a De Novo Review for your medical device or need assistance with any aspect of the regulatory process, please get in touch with Fang Consulting to learn more about our services and how we can help you bring your device to market.

Need a project completed? We have an expert for that.
Contact Us