In vitro diagnostic medical devices (IVDs) are essential tools for diagnosing, monitoring, and treating diseases.
IN VITRO DIAGNOSTIC REGULATION

Expert Support Navigating In Vitro Diagnostic Regulation (IVDR)
In vitro diagnostic medical devices (IVDs) are essential tools for diagnosing, monitoring, and treating diseases. They are widely used in healthcare systems worldwide and are critical in improving patient outcomes. In the European Union (EU), IVDs are regulated by the In Vitro Diagnostic Regulation (IVDR). The IVDR is a new regulation that replaces the EU’s current In Vitro Diagnostic Directive (IVDD) and sets out new rules for designing, manufacturing, and distributing IVDs in the EU.
What’s the difference between IVDD and IVDR?
The IVDR was introduced to address some of the shortcomings of the IVDD and to ensure that IVDs placed on the market are safe, effective, and of high quality. The regulation establishes a greater emphasis on clinical evidence, risk management, and post-market surveillance. IVD manufacturers must provide more extensive and detailed clinical data than was required under the IVDD. This regulation means that manufacturers must perform clinical studies to demonstrate the safety and efficacy of their IVDs before they can sell on the market.
The new risk-based classification system is one of the most significant changes introduced by the IVDR. The IVDR establishes four risk classes based on both patient and public health risks:
- Class A – Low patient and public health risk
- General lab instruments
- Specimen receptacles
- Class B – Moderate patient risk and/or low public health risk
- Some self-test products such as pregnancy, fertility, cholesterol and devices that detect glucose
- Class C – High patient risk and/or moderate public health risk
- Cancer tests, genetic tests, congenital screening of embryos, fetuses or new-born babies
- Class D – High patient risk and high public health risk
- High-risk diseases, blood screening of blood components, determining the infectious load of a life-threatening disease
This system determines the level of scrutiny and oversight that a particular IVD will receive. The classification is based on the device’s intended use, the type of sample used, and the risk to patients, public health, and other factors. The risk-based classification system means that higher-risk IVDs will require more stringent regulatory oversight and must undergo a conformity assessment by a notified body before they can be placed on the market. The regulation also strengthens the post-market surveillance requirements for IVDs, requiring manufacturers to monitor the performance of their devices and report any adverse events or incidents.
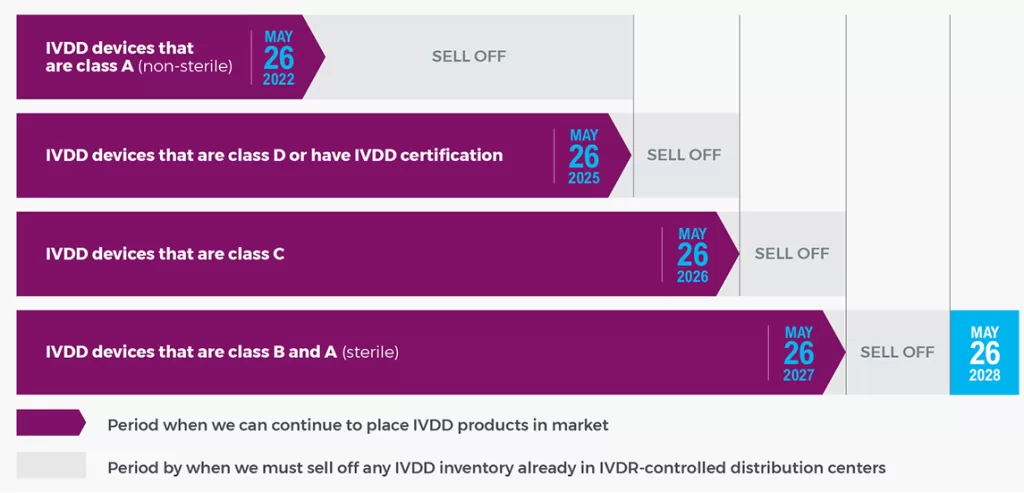
IVDR Implementation Timeline
We’re Here to Help
Compliance with the IVDR can be challenging, particularly for small and medium-sized enterprises needing more resources or expertise. This challenge is where Fang Consulting comes in. We understand the complexities of the IVDR and can help medical device companies navigate the regulatory landscape. Our team of regulatory experts can provide guidance on the requirements for data submission, clinical evaluations, and risk management. We can also assist with preparing and submitting the necessary documentation for CE marking and regulatory compliance.
Our services include regulatory strategy development, preparation of technical documentation, clinical evaluation and study management, risk management and assessment, post-market surveillance and vigilance, and notified body selection and management. We work closely with our clients to understand their specific needs and challenges and tailor our services accordingly. We can help medical device companies of all sizes, from start-ups to established organizations, with their regulatory compliance needs.
We understand that navigating the regulatory landscape can be challenging, particularly in the current environment of increased regulatory scrutiny. Our team has extensive experience in regulatory affairs, and we stay current with the latest regulatory developments, including the IVDR. We can help our clients bring safe and effective IVDs to market and improve patient outcomes.
Compliance with the IVDR is essential for medical device companies that want to place their IVDs on the European market. Our regulatory experts provide guidance on the requirements for data submission, clinical evaluations, and risk management. We can also assist with preparing and submitting the necessary documentation for CE marking and regulatory compliance. Contact us today to learn how we can help you achieve regulatory compliance with the IVDR and bring your IVD to the European market.
IN VITRO DIAGNOSTIC REGULATION
In vitro diagnostic medical devices (IVDs) are essential tools for diagnosing, monitoring, and treating diseases.

How can we help?
Expert Support Navigating In Vitro Diagnostic Regulation (IVDR)
In vitro diagnostic medical devices (IVDs) are essential tools for diagnosing, monitoring, and treating diseases. They are widely used in healthcare systems worldwide and are critical in improving patient outcomes. In the European Union (EU), IVDs are regulated by the In Vitro Diagnostic Regulation (IVDR). The IVDR is a new regulation that replaces the EU’s current In Vitro Diagnostic Directive (IVDD) and sets out new rules for designing, manufacturing, and distributing IVDs in the EU.
What’s the difference between IVDD and IVDR?
The IVDR was introduced to address some of the shortcomings of the IVDD and to ensure that IVDs placed on the market are safe, effective, and of high quality. The regulation establishes a greater emphasis on clinical evidence, risk management, and post-market surveillance. IVD manufacturers must provide more extensive and detailed clinical data than was required under the IVDD. This regulation means that manufacturers must perform clinical studies to demonstrate the safety and efficacy of their IVDs before they can sell on the market.
The new risk-based classification system is one of the most significant changes introduced by the IVDR. The IVDR establishes four risk classes based on both patient and public health risks:
- Class A – Low patient and public health risk
- General lab instruments
- Specimen receptacles
- Class B – Moderate patient risk and/or low public health risk
- Some self-test products such as pregnancy, fertility, cholesterol and devices that detect glucose
- Class C – High patient risk and/or moderate public health risk
- Cancer tests, genetic tests, congenital screening of embryos, fetuses or new-born babies
- Class D – High patient risk and high public health risk
- High-risk diseases, blood screening of blood components, determining the infectious load of a life-threatening disease
This system determines the level of scrutiny and oversight that a particular IVD will receive. The classification is based on the device’s intended use, the type of sample used, and the risk to patients, public health, and other factors. The risk-based classification system means that higher-risk IVDs will require more stringent regulatory oversight and must undergo a conformity assessment by a notified body before they can be placed on the market. The regulation also strengthens the post-market surveillance requirements for IVDs, requiring manufacturers to monitor the performance of their devices and report any adverse events or incidents.
IVDR Implementation Timeline
We’re Here to Help
Compliance with the IVDR can be challenging, particularly for small and medium-sized enterprises needing more resources or expertise. This challenge is where Fang Consulting comes in. We understand the complexities of the IVDR and can help medical device companies navigate the regulatory landscape. Our team of regulatory experts can provide guidance on the requirements for data submission, clinical evaluations, and risk management. We can also assist with preparing and submitting the necessary documentation for CE marking and regulatory compliance.
Our services include regulatory strategy development, preparation of technical documentation, clinical evaluation and study management, risk management and assessment, post-market surveillance and vigilance, and notified body selection and management. We work closely with our clients to understand their specific needs and challenges and tailor our services accordingly. We can help medical device companies of all sizes, from start-ups to established organizations, with their regulatory compliance needs.
We understand that navigating the regulatory landscape can be challenging, particularly in the current environment of increased regulatory scrutiny. Our team has extensive experience in regulatory affairs, and we stay current with the latest regulatory developments, including the IVDR. We can help our clients bring safe and effective IVDs to market and improve patient outcomes.
Compliance with the IVDR is essential for medical device companies that want to place their IVDs on the European market. Our regulatory experts provide guidance on the requirements for data submission, clinical evaluations, and risk management. We can also assist with preparing and submitting the necessary documentation for CE marking and regulatory compliance. Contact us today to learn how we can help you achieve regulatory compliance with the IVDR and bring your IVD to the European market.

Need a project completed? We have an expert for that.
Contact Us