FDA Premarket Approval (PMA)
Walk through the FDA Premarket Approval process with Fang Consulting – your trusted medical device regulatory affairs partner.
Walk through the FDA Premarket Approval process with Fang Consulting – your trusted medical device regulatory affairs partner.
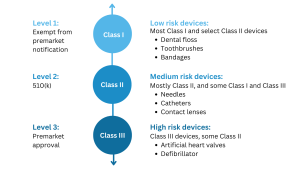
If you intend to introduce a new medical device to the market in the United States, the first step is to determine its regulatory classification. Most medical devices overseen by the FDA are classified as either Class I or Class II, and their marketing requires a 510(k) premarket notification or a simple registration if exempt from 510(k) requirements. However, if your device plays a critical role in sustaining or supporting life, is implanted in the body, or poses a “potential unreasonable risk of illness or injury,” it is likely classified as a Class III device. For such devices, obtaining Premarket Approval (PMA) from the FDA is necessary before they can be marketed in the United States. Additionally, devices that are considered novel, meaning there are no existing substantially equivalent devices, are automatically classified as Class III. However, novel devices with a lower risk profile may be eligible for the De Novo process instead of the PMA. It is important to note that only around 10% of the medical devices regulated by the FDA fall into the Class III category.
Premarket Approval (PMA) is the FDA process of scientific and regulatory review to evaluate the safety and effectiveness of Class III medical devices.
According to the FDA, Class III medical devices are:
Given the high risk associated with these devices, the FDA has determined that general and special controls are inadequate to ensure their safety and effectiveness. Thus, Class III devices necessitate premarket approval (PMA) application under section 515 of the FD&C Act to attain marketing approval.
PMA devices frequently introduce novel concepts, and many fall outside the types of medical devices marketed before the Medical Device Amendments. As a result, these devices may lack a regulatory classification in the CFR. Under such circumstances, the product classification database will only provide the device type name and product code.
The PMA process involves a rigorous review of scientific and clinical data and can take years to complete. The review of a premarket approval application (PMA) is a four-step review procedure consisting of the following:
The FDA has published the following guidance to help industry navigate the PMA process:
Premarket Approval (PMA) | FDA
It is in the manufacturer’s best interest to seek the support of a consulting group, such as Fang Consulting, to ensure all documentation is scientifically sound and well-organized prior to submission.
Fang Consulting has a team of experts with extensive experience in FDA PMA submissions. We provide a range of services to help manufacturers prepare and submit their PMA applications, including:
We aim to help medical device manufacturers navigate the complex FDA approval process and achieve successful PMA submissions. We understand that the PMA process can be daunting, but with our expertise and support, you can feel confident in achieving successful FDA approval. Contact us today to learn more about our PMA submission services and how we can help you navigate the FDA approval process.