EU IVDR Consulting
Our consulting services will make
sure your company is fully prepared
for the new regulatory requirements.
Our consulting services will make
sure your company is fully prepared
for the new regulatory requirements.
The In Vitro Diagnostics Regulation (IVDR) requires most IVD medical device manufacturers to obtain a CE Mark through a EU Notified Body. Our consulting services will make sure your company is fully prepared for the new regulatory requirements.
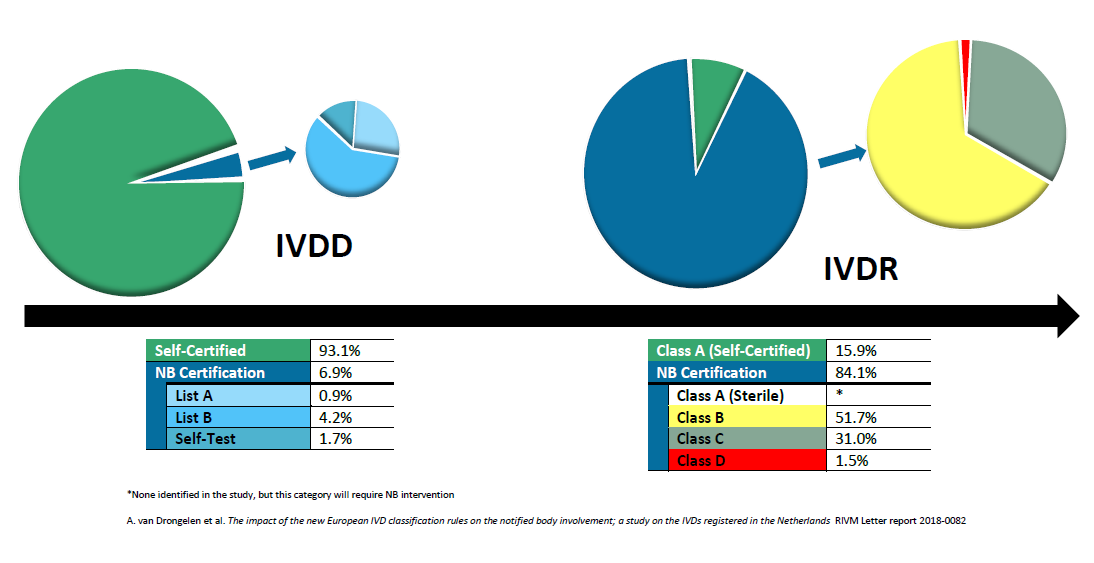 In Vitro Diagnostics (IVDs) are undergoing a huge change to certification in Europe. Currently, most IVDs are self-certified, and do not require the involvement of a Notified Body. IVDR is expected to completely change this with nearly 85% of all IVDs requiring Notified Body involvement, leaving only 15% of IVDs eligible for self-certification.
In Vitro Diagnostics (IVDs) are undergoing a huge change to certification in Europe. Currently, most IVDs are self-certified, and do not require the involvement of a Notified Body. IVDR is expected to completely change this with nearly 85% of all IVDs requiring Notified Body involvement, leaving only 15% of IVDs eligible for self-certification.
*https://www.rivm.nl/bibliotheek/rapporten/2018-0082.html
IVDR regulation enforcement will begin on May 26, 2022.
Fang Consulting is equipped with the knowledge to provide regulatory consulting services and navigate the new EU IVDR regulation requirements to gain market approval. Our consultants will educate your team on why and how the new regulations will likely impact your IVD products. We understand the impact of the new IVDR requirements, including technical documentation and post-market surveillance expectations. We also understand the needs of manufacturers with increased Notified Body involvement in obtaining CE Mark, which is new to many IVDs under the new IVDR.
No. There are zero indications that the timeline will be extended.
Unless you currently hold a valid IVDD CE certificate issued by a Notified Body that expires later, you must comply with IVDR by May 26, 2022.
Most likely no. About 85% of devices will now require a Notified Body to continue sales in the EU.
The legal manufacturer classifies the device. Your Notified Body will review the classification and rationale during the IVDR application process. Discrepancies between your determination and that of the Notified Body can add significant time to your certification, so correct classification with thorough rationale is essential.
Maybe. There is no public database of applications and ETAs. Contacting your current NB is the only way to get accurate information on your specific situation.
No, if IVDR requires Notified Body certification then your product must conform to IVDR by May 26, 2022 to place your product on the market after that date.
The transition period (May 26, 2022 – May 26, 2024) applies to products currently IVDD certified by a Notified Body with a valid certificate that are allowed to be placed on the EU market. This does not apply to products that are self-certified under IVDD.
All certificates to IVDD are void on May 27, 2024 and those devices can no longer be placed on the market.
One option is to start collecting Post Market Clinical Follow-Up data now so you can leverage it for your submission. This can be difficult during COVID-19 restrictions, so consider PMCF surveys. They are a valid method and can be performed remotely without site visits or patient access.
Very few companies have the data required to submit for IVDR because exponentially more information is required compared to IVDD. Most companies also do not have a Notified Body under contract for IVDR.
Based on typical remediation timelines, start now to be certified in time to keep your products on the EU market.
No. The current version of ISO 13485 does not cover all the required elements of IVDR so you will need to make some additions to meet those requirements. All products (except Class A non-sterile) require a Notified Body audit of your quality system to IVDR elements.